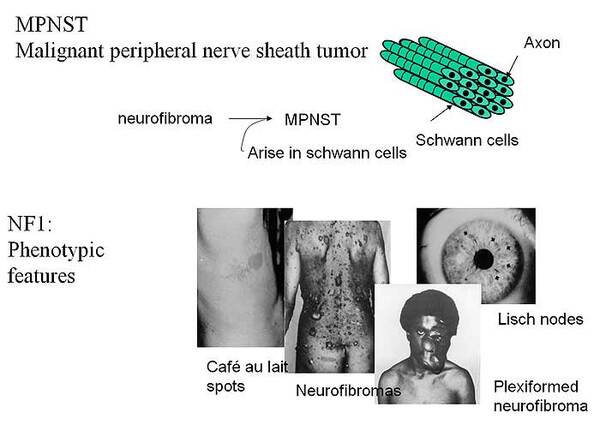
The incidence of malignant peripheral nerve sheath tumour (MPNST) is low in the general population, but individuals with the hereditary disease neurofibromatosis type 1 (NF1) are at greatly increased risk to develop this cancer. About two-thirds of all MPNSTs arise in neurofibromas, often of the plexiform type and in the setting of NF1. Early detection and the differential diagnosis of MPNST remain medical challenges. MPNSTs are highly aggressive and the patients have a poor prognosis.
Development of targeted therapies based on recent advances in molecular biology have been raised as key strategy in the International consensus statement on MPNST in Neurofibromatosis 1.
The NF1 tumor suppressor gene maps to chromosome band 17q11.2, and encodes the neurofibromin protein which function as a suppressor of RAS mediated signaling. Neurofibromin is expressed in the Schwann cells from which the MPNSTs are believed to originate. Individuals with NF1 carry a germline mutation in this gene. Several studies have shown loss of chromosome arm 17q sequences including the NF1 locus. Thus, a complete inactivation of NF1 is assumed to contribute to development of MPNSTs. Potential therapies include inactivation of RAS by preventing post-translational modifications like farnesylation or to block downstream targets of the RAS mitogenic signaling pathway. Somatic mutations in NF1 are also reported in benign neurofibromas, showing that additional genetic events beside inactivation of NF1 are necessary for malignant transformation.
In contrast to several soft tissue sarcomas no pathognomonic structural aberration has been found in MPNST that rather display complex karyotypes. However, recurrent chromosomal alterations have been identified in MPNSTs, including loss of 9p21, shown to reflect downregulated expression of CDKN2A (p16). Further, we have recently shown that frequent gain of 17q (distal to the map position of NF1) partly explain the upregulation of TOP2A, which is typically present in MPNST from patients with poor disease outcome (Skotheim et al., 2003). It has also been suggested that loss of TP53 function contribute to MPNST progression, but biallelic inactivation is rare in these tumours.
Publications: Storlazzi C, Brekke H, Mandahl N, Brosjo O, Smeland S, Lothe R, and Mertens F (2006). Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J.Pathol. .
Ågesen TH, Flørenes VA, Molenaar I, Lind GE, Berner JM, Plaat BEC, Komdeur R, Myklebost O, van den Berg E, and Lothe RA (2005). Expression patterns of cell cycle components in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. J.Neuropathol.Exp.Neurol. 64(1): 74-81.
Skotheim RI, Kallioniemi A, Bjerkhagen B, Mertens F, Brekke HR, MonniO, Mousses S, Mandahl N, Sæter G, Nesland JM, Smeland S, KallioniemiO-P, and Lothe RA (2003). Topoisomerase IIa is upregulated in malignantperipheral nerve sheath tumors and associated with clinical outcome.J.Clin.Oncol., 21(24): 4586-4591.
Lothe RA, Smith-Sørensen B, Hektoen M, Stenwig AE, Mandahl N, Sæter G,and Mertens F (2001). Biallelic inactivation of TP53 rarely contributesto the development of malignant peripheral nerve sheath tumors. GenesChromosomes.Cancer, 30(2): 202-206.
Berner JM, Sørlie T, Mertens F, Henriksen J, Sæter G, Mandahl N,Brøgger A, Myklebost O, and Lothe RA (1999). Chromosome band 9p21 isfrequently altered in malignant peripheral nerve sheath tumors: studiesof CDKN2A and other genes of the pRB pathway. Genes Chromosomes.Cancer,26(2): 151-160.
Lothe RA, Karhu R, Mandahl N, Mertens F, Sæter G, Heim S, Børresen-DaleAL, and Kallioniemi OP (1996). Gain of 17q24-qter detected bycomparative genomic hybridization in malignant tumors from patientswith von Recklinghausen's neurofibromatosis. Cancer Res., 56(20):4778-4781.
Lothe RA, Slettan A, Sæter G, Brøgger A, Børresen AL, and Nesland JM(1995). Alterations at chromosome 17 loci in peripheral nerve sheathtumors. J.Neuropathol.Exp.Neurol., 54(1): 65-73.
Lothe RA, Sæter G, Danielsen HE, Stenwig AE, Høyheim B, O'Connell P,and Børresen AL (1993). Genetic alterations in a malignant schwannomafrom a patient with neurofibromatosis (NF1). Pathol.Res.Pract., 189(4):465-471.
Moosavi SH, Kryeziu K, Eilertsen IA, Nunes L, Hektoen M, Niederdorfer B, Reims HM, Syversveen T, Grut H, Dueland S, Line PD, Lothe RA, Sveen A(2025) Molecular prognostic factors for liver transplantation of unresectable metastatic colorectal cancer Br J Surg, 112(4) DOI 10.1093/bjs/znaf072, PubMed 40235343
Ugalde-Morales E, Wilf R, Pluta J, Ploner A, Fan M, Damra M, Aben KK, Anson-Cartwright L, Chen C, Cortessis VK, Daneshmand S, Ferlin A, Gamulin M, Gietema JA, Gonzalez-Niera A, Grotmol T, Hamilton RJ, Harland M, Haugen TB, Hauser R, Hildebrandt MAT, Karlsson R, Kiemeney LA, Kim J, Lessel Det al.(2025) Identification of genes associated with testicular germ cell tumor susceptibility through a transcriptome-wide association study Am J Hum Genet, 112(3), 630-643 DOI 10.1016/j.ajhg.2025.01.022, PubMed 39999848
Birgisdóttir A, Myklebust TÅ, Bjørnslett M, Rognlien VW, Paulsen T, Dørum A(2024) BRCA mutation testing and association with oncologic outcome and incidence of ovarian cancer in Norway Int J Gynecol Cancer, 35(2), 100029 DOI 10.1016/j.ijgc.2024.100029, PubMed 39971428


 Storlazzi C, Brekke H, Mandahl N, Brosjo O, Smeland S, Lothe R, and Mertens F (2006). Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J.Pathol. .
Storlazzi C, Brekke H, Mandahl N, Brosjo O, Smeland S, Lothe R, and Mertens F (2006). Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J.Pathol. .