ADOPTIVE T-CELL THERAPY (CAR)
Ideally, efficient tumour-specific effector and memory T cells can be induced by therapeutic vaccination. Nevertheless, in certain cases, active immunization is difficult due to the lack of an effective endogenous T-cell repertoire against the tumour antigens targeted.
T cells can recognize peptides derived from all parts of the cell, including nuclear proteins, which greatly expands the number of potential targets in the tumour cells and offers a large number of new therapeutic opportunities.
Adoptive cell therapy (ACT) involves the administration of large number of highly selected cells with high avidity for tumour antigens.
T cells can be programmed and activated ex vivo to exhibit anti-tumour functions. These cells occur naturally in cancer patients, but are inhibited by numerous immunosuppressive mechanisms in vivo. Adoptive transfer of tumour-specific T cells from tumour infiltrating lymphocytes (TIL) expanded in vitro has already been shown to induce objective cancer regression in patients with metastatic melanoma. A limitation of this approach is the requirement for pre-existing tumour-reactive cells that can be expanded ex vivo and TILs can only be reliably grown from patients with a very limited number of cancer types and mainly melanoma.
In the absence of an adequate level of endogenous tumour-reactive T-cell response, recent clinical studies have shown that it is feasible to compensate for this by engineering a tumour-specific T-cell repertoire by the transfer of genes encoding TCRs or chimeric antigen receptors (CARs). Both of these methods are developed in-house (see below).
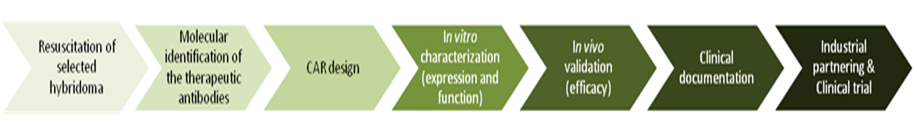
CAR (PI: S. Wälchli/E. Smeland at ICR):
Antibodies are produced by B cells and released into blood and body fluids to block pathogens. They are highly specific and show a high affinity to their antigen (e.g. a part of a protein). By genetic modification, the antigen recognition part of the antibody, the variable part (Fv), can be minimized to a small unit (single chain Fv, scFv) that conserves the specificity and the affinity of the original antibody. Twenty five years ago, it was reported that the fusion of a scFv to the signaling domain of the TcR (CD3ζ) could stimulate the expressing T cell upon antigen binding (Eshar et al, 1993). This type of constructs was further named CAR, for chimeric antigen receptor. Since then, the hunt for extracellular cancer markers has exploded and several targets were defined, mainly for B-cell malignancies. CD19, CD20, and CD22 CARs are nowadays in development or already in advanced clinical trials with promising outcomes.
The Radium Hospital possesses a large collection of hybridomas, each producing monoclonal antibodies. Some of these antibodies have already been used in clinical trials, but never tested as CARs. We therefore aim at designing CARs targeted against unexploited antigens.
Funding from the Norwegian Cancer Society and Helse Sør-Øst
Current CAR projects:
Ongoing